
“SAMMI provides accurate and comprehensive large molecule analysis for quadrupoles”
Cerno introduces an entirely new approach for analyzing any large molecule including peptides, oligos, and proteins, SAMMI, for Spectrally Accurate Modeling of Multiply charged Ions. SAMMI uses a direct analytical model based on first principles. It will analyze each and every ion of a multiply charged molecule to provide not only the neutral mass, but also the abundance distribution across all the charge states.
With SAMMI, you visually observe the results in a simple, intuitive, and confident fashion. Since it is based on a robust, direct analytical model, there are no parameters to tweak, SAMMI is both simple and reliable to use. SAMMI also easily accounts for modifications and adducts and not only determines the accurate neutral mass of the unknown molecule, but also can accurately confirm the sequence/formula of the molecule as well as its adducts, modifications, impurities, and isotope labels.
SAMMI relies on TrueCal™, Cerno’s patented MS calibration method which dramatically improves Spectral Accuracy to allow for the most accurate modeling of the mass spectral profiles for any given ion, regardless of the charge state. It dramatically improves the low resolution MS results to approach the performance from a more expensive high resolution instrument. This makes it perfect not only for research, but for high throughput screening or quality control applications using robust, reliable, and more cost-effective quadrupole instruments. SAMMI works with all large molecules from small peptides and large proteins, oligonucleotides such as RNA of various lengths, and other polymers.
Best of all, SAMMI is free with the purchase or upgrade of Cerno’s award winning MassWorks software.
Download the SAMMI Brochure.
(Click image to enlarge)
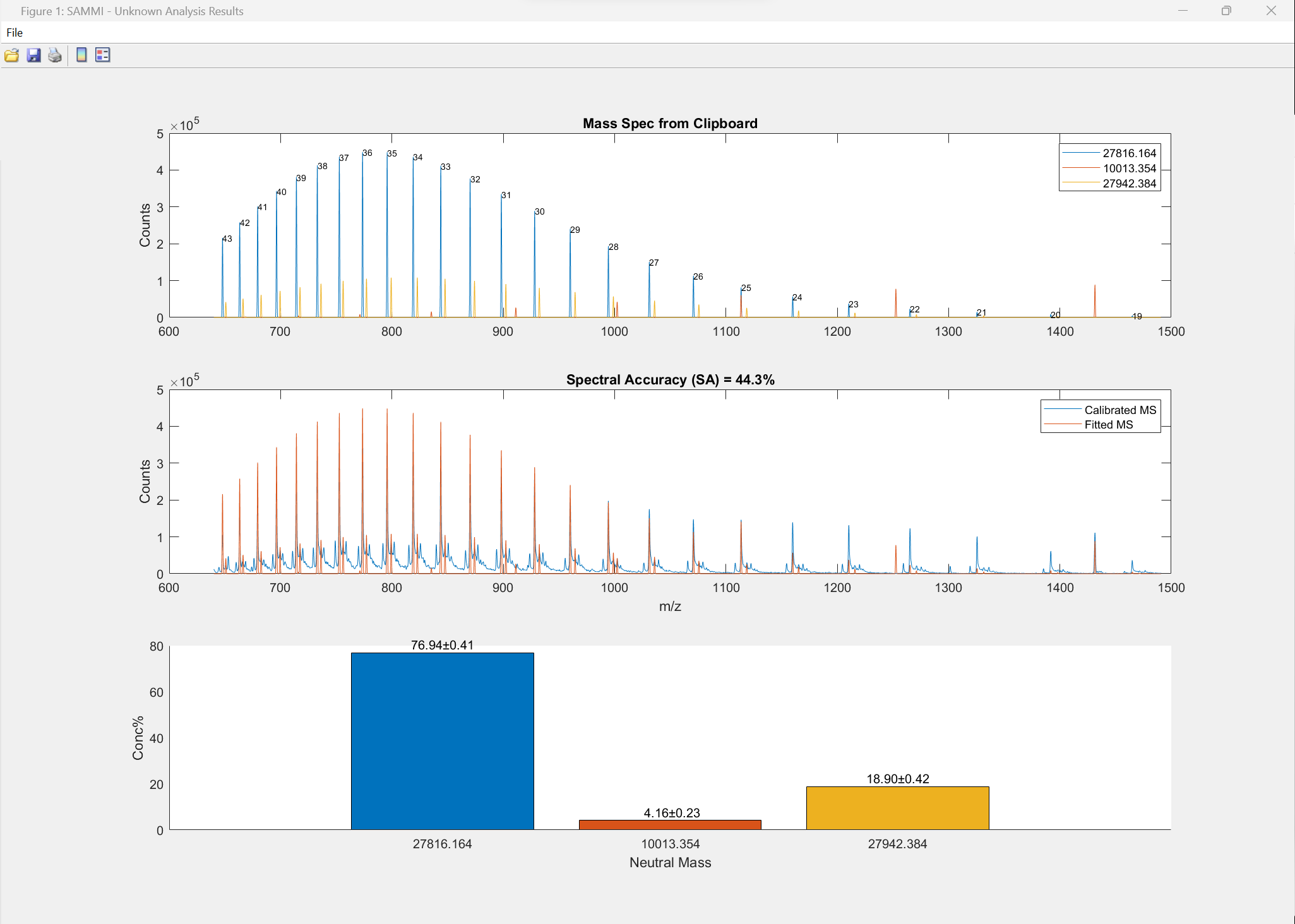
Modified protein analyzed by SAMMI as unknown with 3 components. Actual molecular weight 27816.36, SAMMI estimated MW 27816.15.
Data courtesy of U. of Zurich funded by Swiss National Science Foundation
Key Features
- Uses Cerno’s proven TrueCal™ approach to directly model large molecules with any charge states
- 10-50X improvement in mass accuracy on single or triple quadrupole instruments
- Accurate mass analysis of unknown molecules as well as their proposed adducts, modifications, and impurities along with all their relative concentrations
- Confirm proposed sequences/formula including multiple modifications and/or isotope labels
- No limit to molecule size, large or small
- Provides relative quantitation for all species across all charge states
- Logical, intuitive, and scientifically defendable interpretation of results, i.e., no more “black box
- “Parameter Free” making it very easy-to-use
- Available libraries for direct integration with high throughput workflows using a Python API
- Vendor agnostic
- Patented
Spectral Accuracy for Multiply Charged Large Molecules
Why is Spectral Accuracy so important to the analysis of large molecules? For the analysis of a singly charged small molecule, a single point measurement of mass accuracy can only confirm a single mass location (either monoisotopic or average mass) among a typically wide mass distribution of the molecules’ various isotopes. Spectral Accuracy takes into consideration the entire mass spectral profile including all relevant isotopes and has proven to be a much superior confirmation for the whole molecule. Perhaps more importantly, it can also give a direct indication of any spectral interferences, e.g., the presence of impurities or overlapping ion fragments (e.g. M+ and (M-H)+ or labeled compounds). With spectra that are well calibrated with high spectral accuracy, we can explain and quantitatively account for complex multiply charged spectra along with associated adducts, modifications (e.g. deamidation or deamination), and impurities across all charge states. This is accomplished by simply fitting the respective theoretically calculated mass spectra (true MS) to the calibrated mass spectrum through multiple linear least squares regression. Without the use of Spectral Accuracy and by relying solely on mass accuracy, deamidation / deamination or other modifications can easily escape detection, let alone attain accurate and unbiased quantitation, with either low or high resolution instrumentation.
(Click image to enlarge)
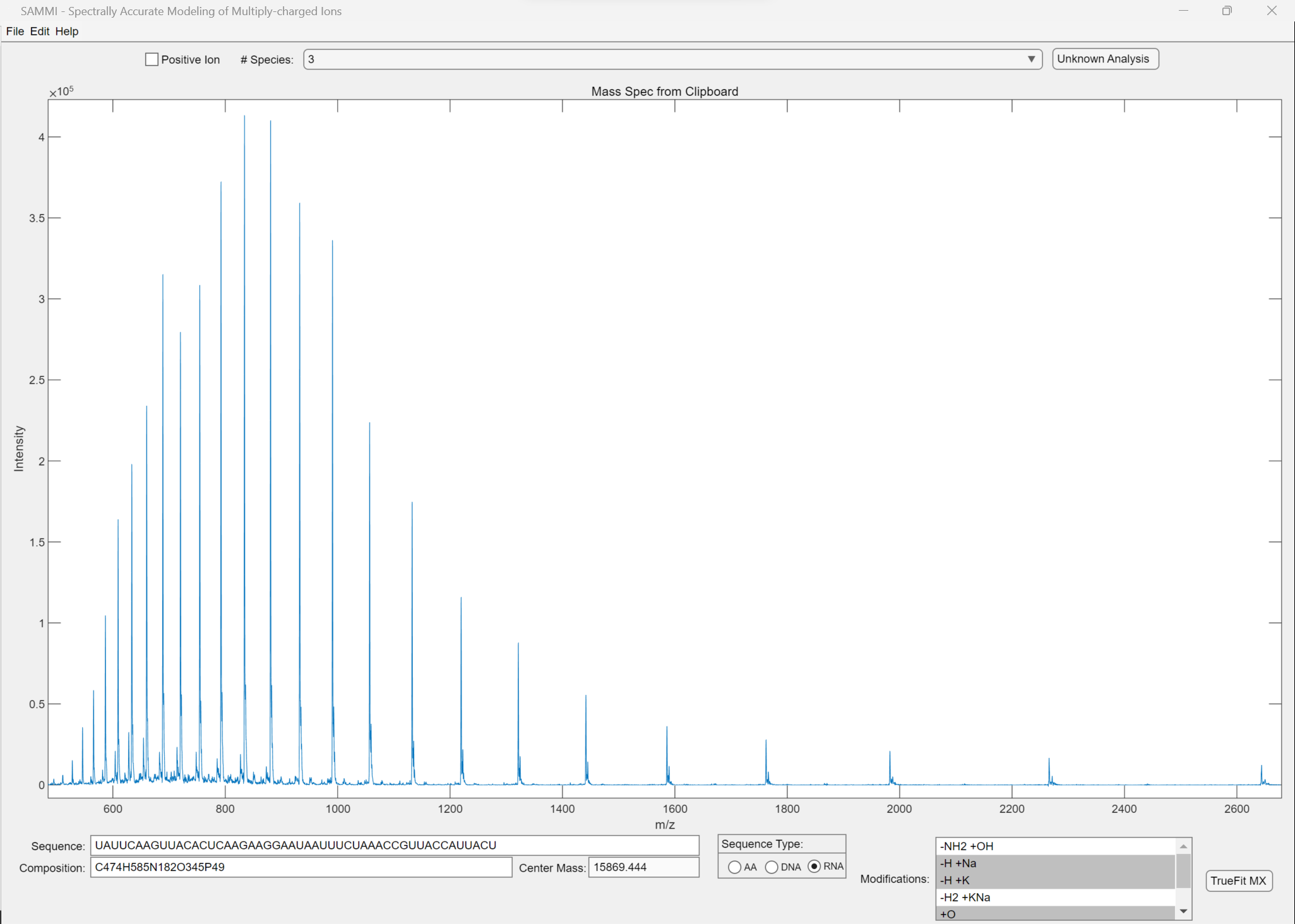
50-nt RNA sample calibrated data run on single quad. Note the ability to include modifications in the analysis (lower right). If the sequence is known, it can be entered (lower left) and the formula and average mass is calculated. The proposed formula can be fitted against the calibrated run data to confirm the correct sequence.
(Click image to enlarge)
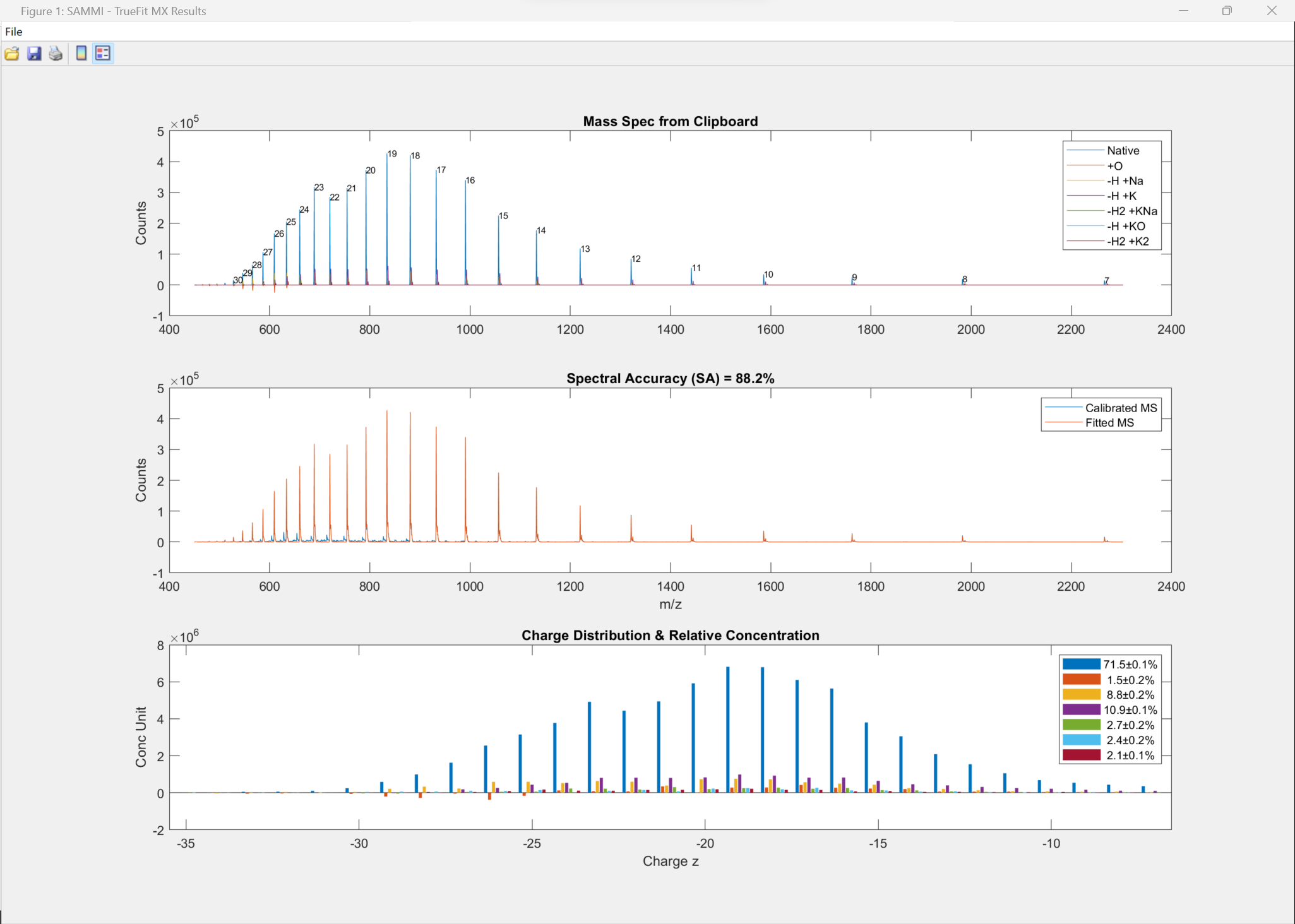
50-nt RNA analyzed against the proposed sequence along with 6 other components (adducts and modifications, top). The excellent spectral accuracy of almost 90% (middle) confirms the compound, adducts and modifications along with a detailed the quantitative relative abundance distribution across all charge states (bottom).
(Click image to enlarge)
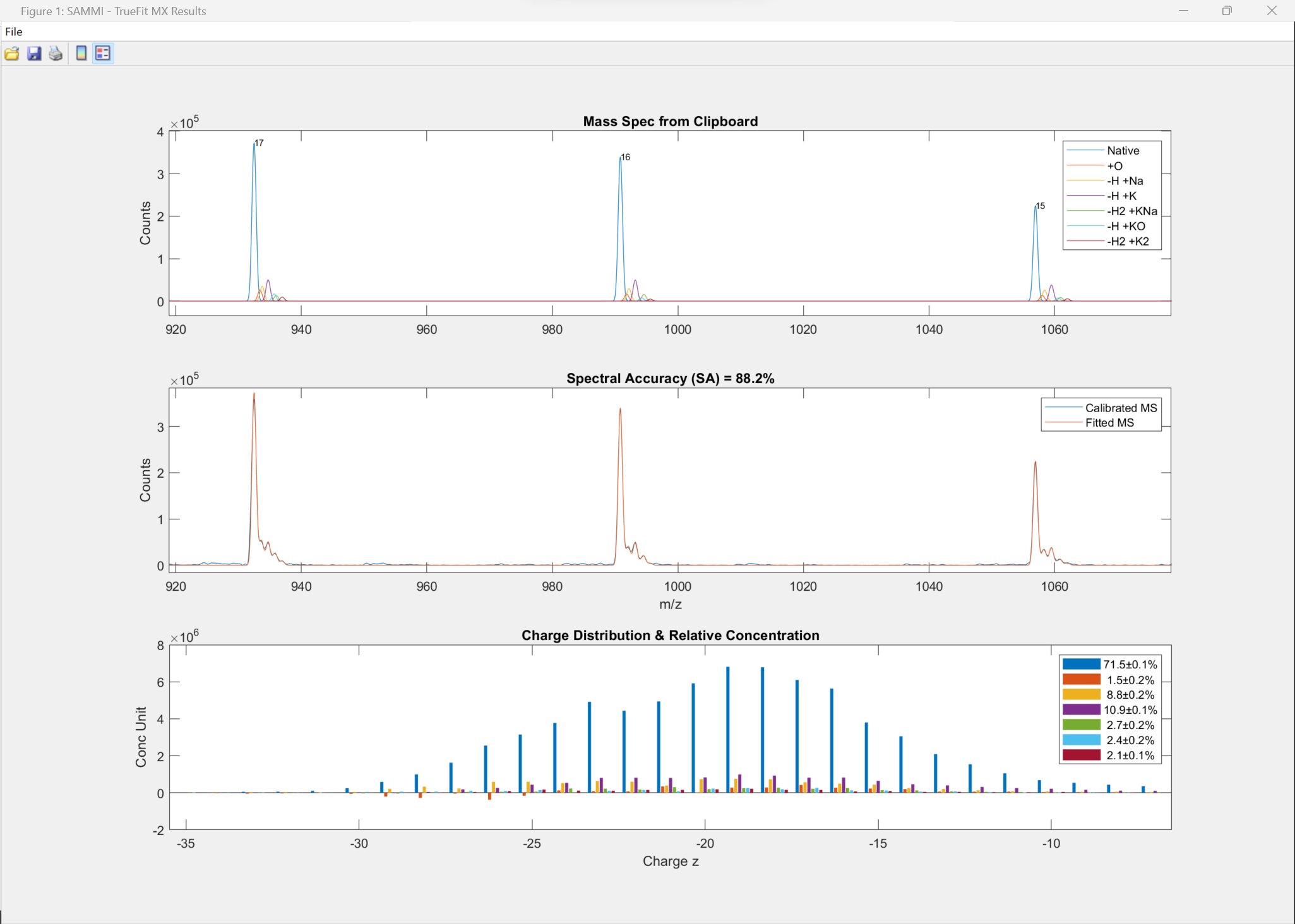
50-nt RNA (zoomed in) shows the calculated component spectra (true MS, top) and the spectral accuracy overlay of the fitted spectra as a linear combination of the native species, its adducts, and modifications against the calibrated mass spectra after TrueCal (middle).